
















Activités
 Réglementer, élaborer des référentiels et inspecter
Réglementer, élaborer des référentiels et inspecter
Mise sur le marché des dispositifs médicaux et dispositifs médicaux de diagnostic in vitro (DM/DMIA/DMDIV)
Nouveau règlement européen pour les dispositifs médicaux
 Base de données EUDAMED pour les dispositifs médicaux : lancement du module destiné à l’enregistrement des opérateurs La Commission européenne a annoncé le lancement le 1er décembre 2020 du premier module de la base de données sur les dispositifs médicaux EUDAMED. Ce module, appelé ACTEURS, destiné aux opérateurs économiques et autorités sanitaires européennes, permettra de préparer l’entrée en vigueur du nouveau règlement européen pour les dispositifs médicaux, dont la date initialement prévue en 2020 a été reportée au 26 mai 2021, du fait de la COVID-19. Nous encourageons les opérateurs établis en France à s’enregistrer sur EUDAMED dès le 1er décembre afin notamment de se familiariser avec la base qui devrait être d’utilisation obligatoire à compter de mai 2022. Lire aussi |
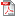 (3275 ko)
(3275 ko)
Le processus de révision complète de la réglementation européenne relative aux dispositifs médicaux s’est achevé en 2017.
L'objectif de cette révision est de renforcer la sécurité sanitaire et d'harmoniser l’application des règles au sein de l’Union européenne.
Elle doit également apporter certaines simplifications, notamment grâce au nouveau système d’information EUDAMED et une meilleure lisibilité des textes législatifs. Elle permettra de mieux accompagner l’innovation.
Cette révision en profondeur de la réglementation a abouti à la publication au JOUE, le 5 mai 2017, d'un nouveau règlement spécifique aux dispositifs médicaux (DM) [UE 2017/745].
 |
|
Lire aussi
- Présentations de la réunion d'information du 13/04/2018 sur le nouveau règlement européen
- Nouveaux règlements européens relatifs aux dispositifs médicaux (29/08/2017) - Point d'information
- Comité d'interface avec les représentants des industries des dispositifs médicaux et dispositifs médicaux de diagnostic in vitro
- Commission européenne
Point marquants du règlement dispositifs médicaux 2017/745
Le nouveau règlement conserve les fondamentaux de la "nouvelle approche" dite de marquage CE, mais il détaille et renforce de nombreuses exigences :
- Le champ d’application est étendu à des dispositifs sans finalité médicale listés en annexe XVI; par contre, les pro-biotiques sont exclus.
-
Des obligations nouvelles sont imposées aux opérateurs économiques
: une personne est chargée de veiller au respect de la réglementation chez le fabricant (et son mandataire)
Des obligations de prudence sont mises en place pour les importateurs et tous les distributeurs (y compris officines et grande distribution).
-
Les organismes notifiés
sont placés sous contrôle européen pour une meilleure harmonisation des pratiques.
Ils répondent désormais à un cahier de charges renforcé en matière de compétence et sont soumis à de nouvelles obligations de procédures (visite inopinée chez les fabricants, contrôles de produits).
- Une véritable régulation du secteur à l'échelon européen est mise en place avec un groupe de coordination des autorités nationales et de nouveaux mécanismes de coopération étroite, notamment pour une surveillance du marché coordonnée.
- Le dispositif de vigilance est amélioré avec la mise en place d’une base européenne des incidents et l’obligation faite aux fabricants sous contrôle des organismes notifiés de produire des résumés périodiques de sécurité (PSUR).
- L’encadrement des investigations cliniques converge avec l'encadrement applicable au médicament pour les essais cliniques.
- L’évaluation avant mise sur le marché est renforcée
- de nouvelles exigences essentielles sont introduites, comme la justification du recours à des substances dangereuses (CMR, PE) ou encore pour la cyber-sécurité.
- pour les DM à base de substances absorbées : de nouvelles procédures de consultation sont créées pour la certification CE auprès d’une autorité compétente en matière de médicament.
-
pour les nouveaux dispositifs implantables : le recours aux investigations cliniques pour l'évaluation devient incontournable.
L’organisme notifié devra consulter un panel d’experts européens sur le dossier clinique des nouveaux DM implantables de classe III.
- Certaines pratiques sont encadrées, comme la production de DM au sein des établissements de santé en la limitant aux besoins médicaux non couverts, ou le retraitement de DM à usage unique pour leur remise sur le marché dans les pays qui voudront autoriser cette pratique.
-
La transparence et la traçabilité sont améliorées
Une véritable base de données européenne des dispositifs médicaux accessible en grande partie au public permettra :- de vérifier facilement la régularité de la présence sur le marché d’un produit donné
- de donner accès à des informations sur les produits, avec un résumé de caractéristiques pour les DM de classe III.
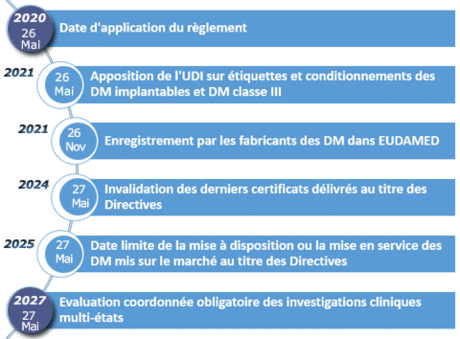
Evaluation, désignation et notification des organismes d'évaluation de la conformité en France
L’ANSM est l’autorité responsable des organismes notifiés au titre de l’article 35 du règlement (UE) 2017/745[1] et du règlement (UE) 2017/746[2] .
En vue de leur désignation en tant qu’organisme notifié, les organismes d'évaluation de la conformité situés en France soumettent un dossier de demande à l’attention du Directeur général de l’ANSM.
| Ces demandes sont envoyées, par voie électronique uniquement, aux adresses suivantes :
|
![]() La demande est instruite par la direction de l’inspection de l’ANSM dans le respect des articles 35 à 43 du règlement 2017/745 et des articles 31 à 39 du règlement 2017/746, ainsi que du document NBOG BPG 2017-1
« Best practice guidance on designation and notification of conformity assessment bodies ».
La demande est instruite par la direction de l’inspection de l’ANSM dans le respect des articles 35 à 43 du règlement 2017/745 et des articles 31 à 39 du règlement 2017/746, ainsi que du document NBOG BPG 2017-1
« Best practice guidance on designation and notification of conformity assessment bodies ».
![]() Les modèles et les guides utilisés lors du processus d’évaluation et de désignation sont ceux adoptés par le groupe de coordination des dispositifs médicaux, disponibles sur le site de la Commission européenne :
Les modèles et les guides utilisés lors du processus d’évaluation et de désignation sont ceux adoptés par le groupe de coordination des dispositifs médicaux, disponibles sur le site de la Commission européenne :
![]() Si l’organisme d’évaluation de la conformité souhaite devenir organisme notifié au titre des deux règlements, il doit verser deux dossiers distincts, qui seront instruits séparément.
Si l’organisme d’évaluation de la conformité souhaite devenir organisme notifié au titre des deux règlements, il doit verser deux dossiers distincts, qui seront instruits séparément.
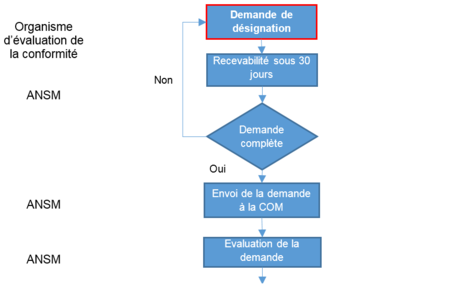
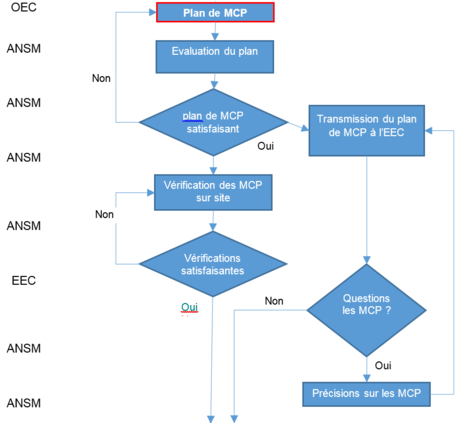
Les étapes d’évaluation et de désignation sont schématisées comme suit :
| Qui | Quoi | Comment |
|
NBOG F 2017-1 (MDR) NBOG F 2017-2 (IVDR) NBOG F 2017-3 (MDR) NBOG F 2017-4 (IVDR) Dossier à envoyer par mail : insmar.dm@ansm.sante.fr (MDR) insmar.div@ansm.sante.fr (IVDR) |
|

|
NBOG F 2017-5 (MDR) NBOG F 2017-6 (IVDR) Composition de l’EEC :
Résumé de l’évaluation conjointe |
|

|
||
|
||

|
Final assessment form (key informations) : |
|
- La désignation de l’organisme notifié prend effet le jour suivant la publication par la Commission européenne de sa notification dans la base de données européenne des organismes notifiés NANDO (https://ec.europa.eu/growth/tools-databases/nando/ ).
- Au-delà de la désignation des organismes notifiés, l’ANSM exerce également une surveillance continue de leurs activités, en lien avec une équipe européenne.
- L’évaluation et la désignation, par l’ANSM, des structures de contrôle et d’évaluation de la conformité des organismes notifiés ne sont sujettes à aucune redevance.
Lire aussi
[1] Règlement (UE) 2017/745 du Parlement européen et du Conseil du 5 avril 2017 relatif aux dispositifs médicaux, modifiant la directive 2001/83/CE, le règlement (CE) n° 178/2002 et le règlement (CE) n° 1223/2009 et abrogeant les directives du Conseil 90/385/CEE et 93/42/CEE] et règlement (UE) 2017/746 [Règlement (UE) 2017/746 DU PARLEMENT EUROPÉEN ET DU CONSEIL du 5 avril 2017 relatif aux dispositifs médicaux de diagnostic in vitro et abrogeant la directive 98/79/CE et la décision 2010/227/UE de la Commission.