
















Dossiers
Médicaments génériques
L'évaluation des médicaments génériques
| L'AMM des médicaments génériques repose sur le processus d’évaluation appliqué à l’ensemble des médicaments  |
|
Comment est évaluée la qualité des médicaments génériques?
Le dossier pharmaceutique doit réunir tous les éléments permettant de justifier de :
- la qualité du médicament (origine et spécifications des matières premières des excipients, caractérisation des impuretés de synthèse et de dégradation, méthodes de fabrication et de contrôle du produit fini),
- la reproductibilité de cette qualité d’un lot à l’autre (validation des méthodes de fabrication et de contrôle)
- du maintien de cette qualité (études de stabilité),
- la similarité du médicament générique au médicament d’origine.
Les dossiers pharmaceutiques des médicaments génériques sont soumis aux mêmes degrés d’exigence et de qualité que ceux des médicaments d'origine.
Lire aussi :
Comment sont évaluées l’efficacité et la sécurité des génériques?
L’efficacité et la sécurité du principe actif contenu dans le médicament générique ont déjà été démontrées pour le médicament d’origine. Les dossiers d’AMM des médicaments génériques n’ont donc pas à produire des essais cliniques.
La preuve de l’efficacité et de la sécurité du médicament générique sont apportées dans le dossier biopharmaceutique par les données de biodisponibilité .
L’évaluation de la qualité de l’étude de bioéquivalence d’un médicament générique fait partie intégrante de l’évaluation d’une demande d’AMM.
Sont notamment pris en compte :
- le nombre de volontaires
- la qualité de la méthode de dosage du principe actif dans le sang
- les méthodes statistiques utilisées pour les calculs
- le nombre de volontaires
- la qualité de la méthode de dosage du principe actif dans le sang
- les méthodes statistiques utilisées pour les calculs
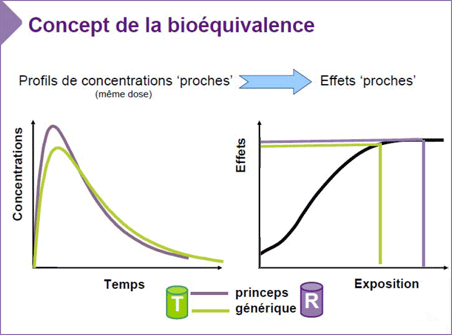 |
| La démonstration de la bioéquivalence entre deux médicaments repose sur la comparaison de leurs biodisponibilités obtenues après l’administration d’une même dose de principe actif par une même voie d’administration. |
Etudes de bioéquivalence
Les études de bioéquivalence permettent de s’assurer que le devenir du principe actif dans l’organisme est équivalent entre le médicament générique et la spécialité d’origine.
Si la bioéquivalence est démontrée, l’exposition à la substance active sera identique entre le médicament générique et le médicament d'origine. Or, si l’exposition est équivalente, cela veut dire que l’efficacité du médicament générique (l’effet pharmacodynamique) sera identique à celle du médicament d'origine.
L’étude est le plus souvent réalisée chez des volontaires sains pour réduire la variabilité interindividuelle inhérente à la maladie.
Elle consiste à comparer, après administration du médicament générique ou du médicament d’origine, la concentration plasmatique du principe actif.
Chaque volontaire reçoit une dose du princeps, puis la même dose du générique, ou inversement. Ainsi chaque volontaire est son propre témoin, c’est un essai croisé. Dans chaque cas, des prélèvements sanguins permettent de mesurer l’évolution de la concentration dans le sang du principe actif au cours du temps.
Les essais de bioéquivalence s’appuient sur la comparaison statistique médicament Générique vs Princeps de 2 paramètres d’exposition systémique au médicament :
- l’aire sous la courbe des concentrations en fonction du temps (AUC pour l’aire sous la courbe ) qui permet d’estimer l’étendue de l’absorption.
- la concentration maximale (Cmax ), mesurée au temps (Tmax ), permettant d’apprécier la vitesse à laquelle le principe actif se retrouve dans l’organisme.
Pour conclure à la bioéquivalence entre les deux médicaments, l’intervalle de confiance du ratio médicament Générique / Princeps doit être entièrement compris dans l’intervalle [80 % ; 125 %].
Cet intervalle de [80 % ; 125 %] a été défini au niveau international en considérant qu’une variation du ratio des moyennes jusqu’à 20 % n’a que peu de conséquences cliniques (efficacité/sécurité).
La FDA a constaté, dans une analyse rétrospective incluant 2 070 études de bioéquivalence soumises entre 1996 et 2007 dans des dossiers de médicaments génériques que la différence des AUC et Cmax entre médicament générique et médicament princeps était en moyenne inférieure à 5 %. (Davit BM et al, Ann Pharmacother, 2009)
Pour les médicaments à marge thérapeutique étroite (concentrations toxiques proches des concentrations efficaces), de faibles variations de dose ou de concentration peuvent être à l’origine de différences importantes d’efficacité ou de tolérance. C’est pourquoi, pour ces médicaments, l’intervalle d’acceptabilité de la bioéquivalence est resserré ([90 % ; 111 %]).
Lire aussi :
- Qu’est-ce que la biodisponibilité et la bioéquivalence? (29/06/2016)
 (275 ko)
(275 ko) - Médicament générique - le point sur la bioéquivalence - Site du Ministère des Affaires sociales et de la Santé
- Des médicaments sûrs et efficaces - Site du Ministère des Affaires sociales et de la Santé